| |||||||||||||||||
|
Stanley B. Prusiner, de la Universidad de California, recibió el Premio Nobel en 1997 por formular la hipótesis sobre la naturaleza priónica de la "encefalopatía espongiforme". Durante el desarrollo de sus investigaciones, acuñó el término "prión" para distinguir el agente infeccioso causante de varias enfermedades neurodegenerativas en mamíferos, como el "scrapie" o "tembladera de las ovejas", la "encefalopatía espongiforme de las vacas", la "enfermedad de Creutzfeldt-Jakob" o el "kuru", estas dos últimas en humanos. La palabra prión deriva de la frase "partícula infecciosa proteínica"; se utiliza para definir a un agente infeccioso que está formado por proteínas y no parece presentar ningún tipo de ácido nucleico (ADN o ARN), como ocurre con el resto de patógenos conocidos (virus, bacterias, etc.) Esta es la hipótesis más aceptada sobre la naturaleza de los priones, habida cuenta que no responden a ningún tratamiento dirigido contra los ácidos nucleicos y sí a los que destruyen proteínas.  Investigaciones posteriores determinaron que el prión que causa estas enfermedades es una proteína que se halla en la membrana de las células normales; suele designarse con la abreviatura "PrP". Dentro de la célula, en su estado normal, el PrP se encuentra en una forma estable ("pN"), que no causa enfermedad alguna. Antes de continuar, queremos incidir en la importancia que tiene la forma espacial, tanto para las proteínas como para otras muchas sustancias que intervienen en la formación de los seres vivos. Así, el reconocimiento enzimático descansa sobre la forma espacial de las enzimas, proteínas que regulan todas las reacciones que se producen en nuestro organismo. La regulación tiene lugar gracias a relaciones del tipo "llave y cerradura". Sólo llaves con una estructura determinada pueden introducirse en una cerradura y abrir la puerta. Estas enzimas reconocen sólo determinadas formas espaciales de otras moléculas, iniciando entonces la cadena de reacciones. Aclarada la importancia de la forma en las sustancias orgánicas, diremos que el PrP puede tomar una forma espacial anómala ("pD"), que es la causante de la enfermedad. Esta forma pD es infecciosa porque, cuando se pone en contacto con la forma normal (pN), la convierte en pD; se trata de un proceso de carácter exponencial, en el que cada pD que se genera transforma a más proteínas normales en anómalas. ¿Qué función tiene el prión en su estado normal? Aunque no se sabe con seguridad, existen indicios que apuntan a que la forma normal del prión (pN) podría intervenir en la protección del cerebro al paso del tiempo, previniendo una temprana muerte celular, como parece ocurrir cuando el prión normal es deficitario o se halla la forma que causa enfermedad. |
||
| Encefalopatías en Humanos |
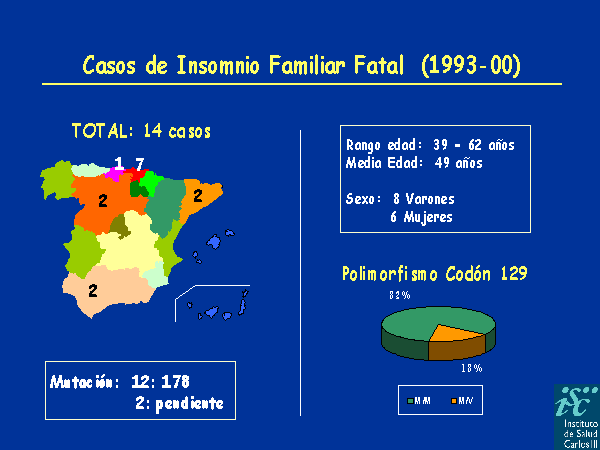
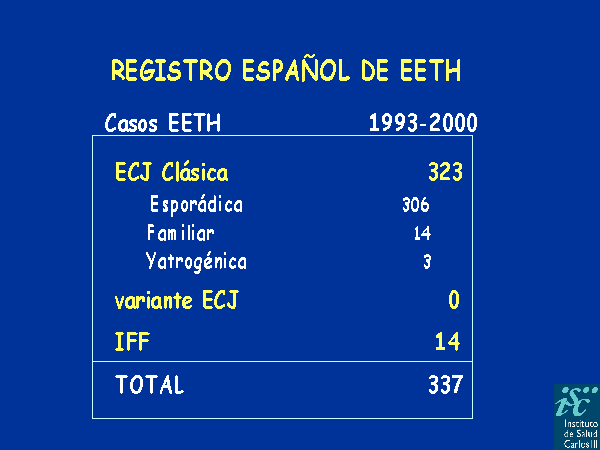
El FFI parece deberse a una deficiencia en la forma priónica normal; se transmite hereditariamente. También son hereditarios el CJ y el GSS que, como la enfermedad anterior, tienen su origen en mutaciones dominantes en el gen para la síntesis de la proteína priónica. Sin embargo, el origen del kuru es distinto; esta enfermedad apareció como una patología epidémica transmisible, similar al CJ, que se extendió por la tribu caníbal de los "Fore", en Papua Nueva Guinea, durante la década de 1950. La enfermedad se extendió entre esta tribu como consecuencia de sus ritos funerarios; durante estas ceremonias, se extraía el cerebro del miembro de la tribu que había muerto, y al que se iba a honrar, y se cocinaba para ser ingerido por sus familiares más cercanos. El análisis de cerebros afectados por la enfermedad de Creutzfeldt-Jakob y por el kuru muestran la misma apariencia espongiforme.
|
|||||||

| Orígenes de la encefalopatía espongiforme bovina (EEB) |
|
Las enfermedades priónicas en los animales también son conocidas desde hace muchos años. La referencia más temprana al scrapie, o tembladera de las ovejas y de las cabras, se remonta a 1732, aunque hasta 1932 no fue reconocida su naturaleza infecciosa. Existen otras enfermedades neurodegenerativas de los mamíferos; la que afecta a las vacas tiene un período de incubación variable, de tres a cinco años; ésta enfermedad es conocida, desde 1920, con el nombre de "encefalopatía espongiforme bovina (EEB), o "mal de las vacas locas". La relación de la EEB con la tembladera de las ovejas tuvo lugar gracias al conocimiento previo de los orígenes y modo de transmisión del kuru en humanos. No parecía improbable la transmisión del prión desde las ovejas a las vacas, dada su cercanía evolutiva. Por ello, se pensó que las vacas podían haberse infectado con el prión anómalo al alimentarlas con harinas que contenían restos de ovejas, obviamente, alguna de ellas enferma de tembladera. Por otra parte, la incorporación de restos animales a la fabricación de harinas comenzó a ser una práctica habitual entre 1978 y 1980. A partir del primer caso de EEB en el Reino Unido, durante 1986, se disparó el número de reses enfermas, por lo que, en 1988, acabaría prohibiéndose el uso de harinas con derivados animales, si bien su exportación a otros países no cesó. Hasta la fecha, en el Reino Unido, se han contabilizado 180.000 casos de vacas locas. La incidencia de esta enfermedad en otros países europeos es mucho menor; en Francia, Suiza, Irlanda y Portugal se han contabilizado un total de 1.300; en Alemania y España los primeros casos han aparecido en el año 2000; y en Italia en enero de 2001. En el Reino unido, al igual que en los países que primero mostraron casos de EEB, la incidencia está disminuyendo tras las medidas adoptadas para su erradicación, si bien, en los países donde no se habían contabilizado casos hasta estos últimos años, la incidencia está en aumento puesto que es ahora cuando esos países están empezando a realizar, de manera masiva, tests de detección de la EEB. |
||
| La variante humana de la encefalopatía espongiforme bovina |
|
Los seres humanos distan evolutivamente mucho de las vacas y las ovejas, por ello se creyó, en principio, improbable que la enfermedad pudiera pasar de una especie a la otra. Dadas las crecientes dudas al respecto, en 1989 la Unión Europea adoptó la primera medida para evitar el consumo humano de carne de vaca contaminada. No obstante, el salto del prión de los bóvidos a los humanos se había producido, provocando una variante, en éstos últimos, de la enfermedad de Creutzfeldt-Jakob (CJ). La forma normal de Creutzfeldt-Jakob aparece, de manera espontánea, en una proporción de un caso por millón y año, afectando a personas de entre 55 y 75 años, en los que se aprecia un progresivo deterioro mental asociado a torpeza, falta de visión, espasmos musculares y otros síntomas neurológicos; durante los últimos estadíos de la enfermedad, el enfermo muestra incapacidad total de hablar y moverse, la muerte sobreviene a los pocos meses de la aparición de los primeros síntomas. En los humanos, la variante del CJ está causada por el mismo agente que la enfermedad bovina y puede afectar a enfermos menores de treinta años. Presenta un proceso más largo, de uno a dos años, así como una sintomatología ligeramente diferente; esta enfermedad parece estar directamente relacionada con la exposición a la EEB, en concreto a través de la ingesta de carne contaminada con el prión. En la actualidad y en el ámbito de la Unión Europea, hay descritos 89 casos confirmados de esta patología en humanos, de los que 85 corresponden al Reino Unido, otros 3 a Francia y 1 a Irlanda. Dado que el período de incubación en humanos ronda los diez años (o más), es posible que este número se incremente en los años siguientes. Del mismo modo, se recomienda que se preste mayor atención a los posibles casos de demencia que cursen un proceso similar en personas mayores, por la posibilidad de que se estén diagnosticando erróneamente casos de encefalopatías.
|
|||||
| Detección de la encefalopatía espongiforme bovina y medidas a tomar para frenar la extensión de estaenfermedad y de la variante de la enfermedad de Creutzfeldt-Jakob |
|
A partir del 1 de enero de 2001 es obligatorio realizar un test post mortem a todos los bóvidos de más de 30 meses que vayan a entrar en la cadena alimentaria, con el fin de descartar los posibles casos de EEB, si bien, en algunos países se ha rebajado la edad para la realización del test a los 24 meses. Otras medidas preventivas tomadas por la Unión Europea han sido la prohibición del uso de harinas, que incorporen carne o hueso de mamíferos para la fabricación de piensos desde 1994; medida que se ha endurecido prohibiendo su uso total para el ganado, a partir del 1 de enero de 2001. Del mismo modo, desde octubre de 2000, se ha ordenado la eliminación del mercado de materiales considerados de alto riesgo (MER) por su elevada concentración de priones, como la médula espinal, el cerebro, los ojos y amígdalas de ternera, oveja y cabra, a los que se ha añadido el intestino, el hueso de espinazo y, últimamente, el chuletón, dado que su corte entraña un posible riesgo de contaminación por verse implicadas algunas de las partes antes comentadas. Los MER no pueden ser utilizados para el consumo animal o humano y tanto éstos, como los animales infectados o susceptibles de padecer la enfermedad por cercanía a otros infectados, así como las harinas de origen animal, han de ser destruidos de alguna de las siguientes formas: incineración directa a temperatura no inferior a 850º C durante, al menos, 2 segundos; transformación en condiciones de 133º C, 3 bares ó 20 mn de presión, en instalaciones controladas que puedan alcanzar esos requerimientos, como centrales eléctricas, cementeras o incineradoras, seguido de la eliminación en vertederos controlados. La leche y sus derivados, así como el sebo y la gelatina son considerados seguros. Entre los materiales no alimentarios susceptibles de producir la transmisión del prión, si bien de riesgo limitado, se encuentran las vacunas, tanto las dirigidas a humanos como las de uso veterinario, así como los cosméticos preparados con material procedente de bovinos.
Para saber más Para saber menos
|
||||
